The research programs in the Stephens lab are focused on understanding the basic mechanisms of pancreatic β-cell function and survival with the long-term goal of developing effective β-cell therapies for treatments of major forms of diabetes. Through this work, we have: i) developed sophisticated imaging platforms to study molecular determinants of proinsulin trafficking -this led to the discovery of ER and Golgi export defects in diabetes models; ii) identified a metabolic redox relay between the mitochondria and ER in β-cells that is necessary for insulin biosynthesis -defects in this relay contribute to the insulin deficiency in Type 2 diabetes; and iii) identified cytokine-directed changes in β‑cell Golgi function that may contribute to T1D pathogenesis.
Pancreatic β-cells produce insulin
Insulin is a peptide hormone uniquely made in pancreatic β-cells that is the only hormone in the body capable of reducing blood glucose levels. In the endoplasmic reticulum (ER), the insulin precursor, proinsulin, is folded and stabilized via the formation of 3 disulfide bonds that are essential for insulin structure. Proinsulin is subsequently trafficked to the Golgi and packaged into storage vesicles, termed dense core secretory granules, where proinsulin is processed via proteolysis to mature insulin. A typical β-cell contains 8000-10,000 mature insulin granules, which occupy roughly 20% of the total β-cell volume. Mature insulin granules can be mobilized for insulin exocytosis upon glucose stimulation. For more information, see our review.
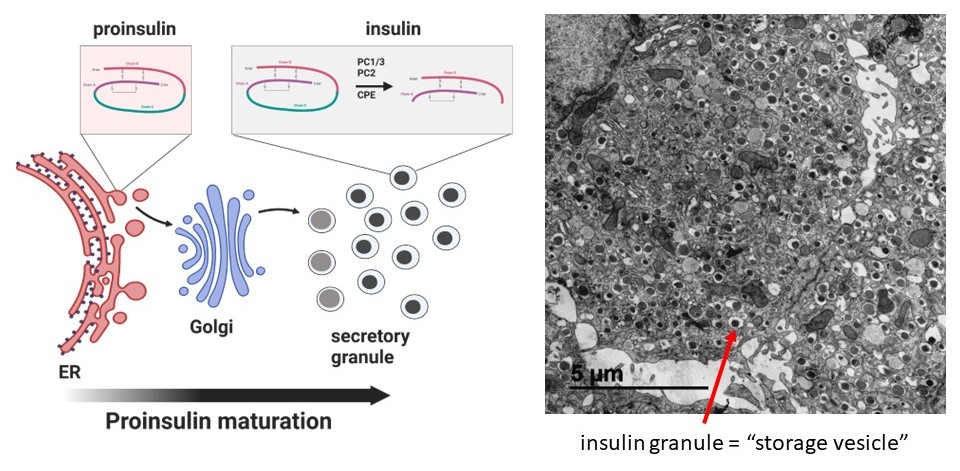
Imaging platforms to study insulin maturation
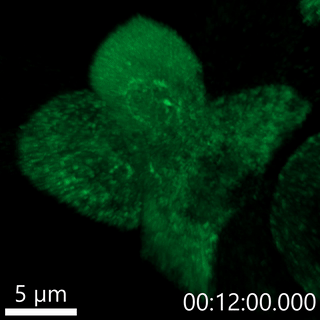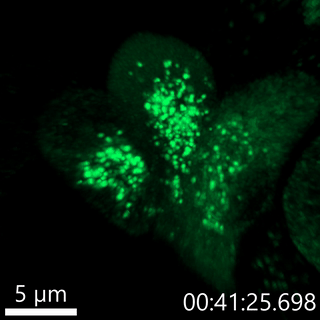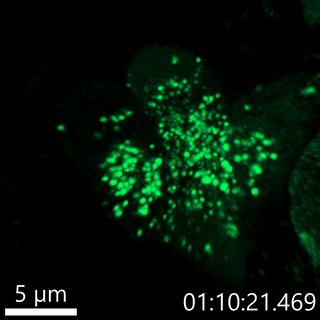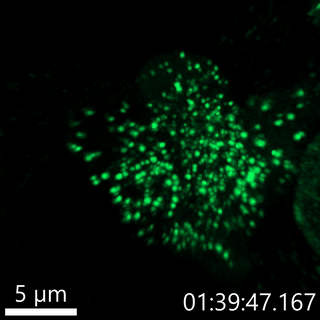
Our lab has developed fluorescent pulse-chase reporters, SNAPtag and RUSH, to identify molecular determinants of proinsulin trafficking. In the movie below, we use the RUSH system to visualize proinsulin trafficking in primary β-cells. We recently identified defects in Golgi export of proinsulin in diabetes models using the RUSH system.
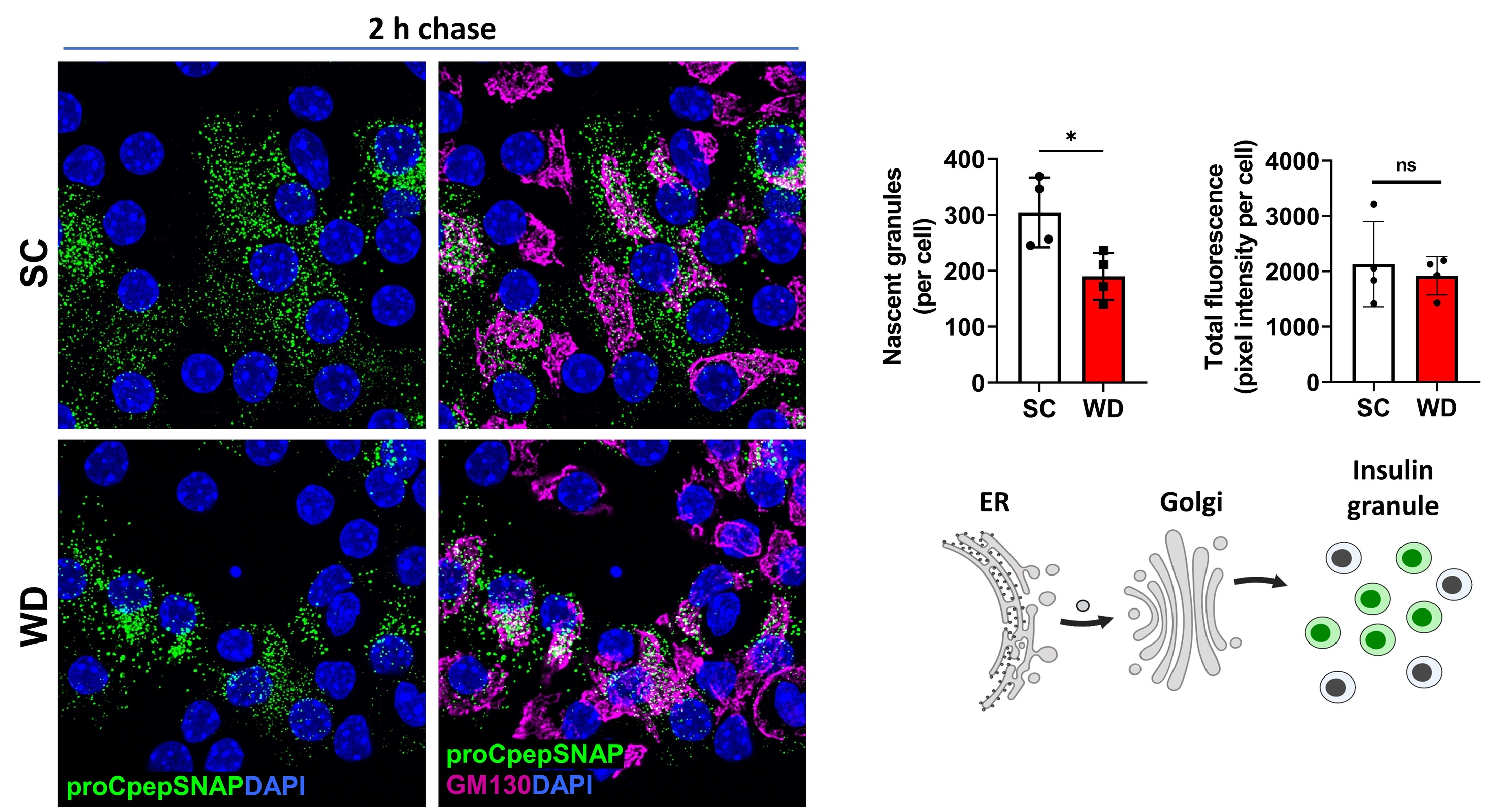
Similarly, SNAPtag has been an invaluable reagent to assess defects in proinsulin trafficking in diabetes models. In the images below, we compared β-cells from mice fed a standard chow (SC) vs. a diabetogenic, western diet (WD; high in sucrose and fat). Here, we observed a large decrease in new insulin granule formation in diabetic WD fed mice. Based on these data, we propose that proinsulin trafficking defects contribute to the insulin deficiency in Type 2 diabetes.
Metabolic control of proinsulin trafficking
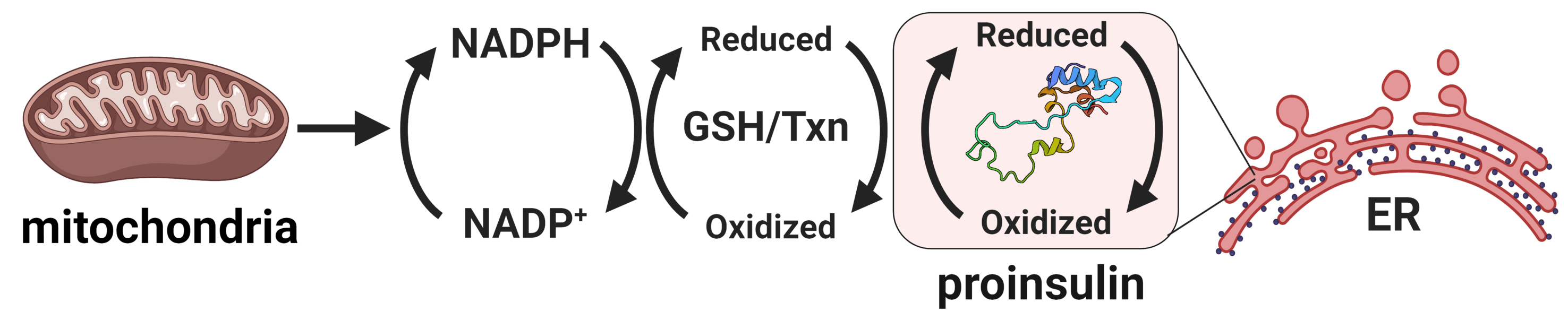
Compelling evidence highlights the central role of islet β-cell failure in the transition from insulin resistance to Type 2 diabetes (T2D). Endoplasmic reticulum (ER) stress and proinsulin misfolding have been heralded as contributing factors to β-cell dysfunction in T2D, yet how ER function becomes compromised is not well understood. Our work has identified altered ER redox homeostasis as a novel mechanism that contributes to insulin granule loss in diabetes. This change in the β-cell's ER redox potential is a substantial burden to proinsulin folding, which ultimately may limit the proinsulin supply available for insulin maturation.
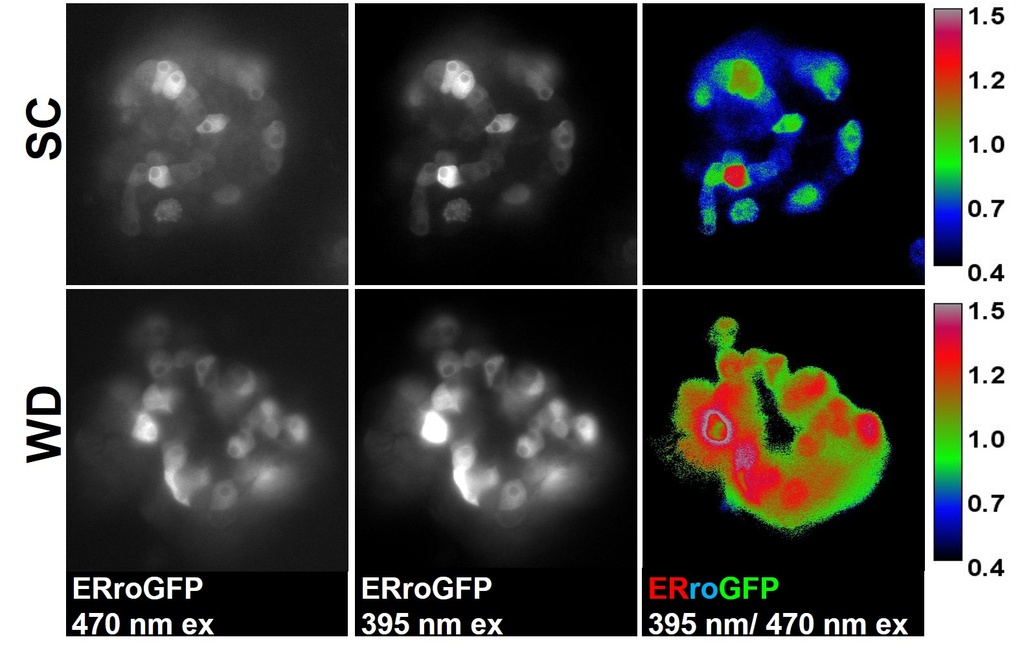
Our recent studies have now identified glucose metabolism as a critical determinant in the redox homeostasis of the ER. Using multiple β-cell models, we show that loss of mitochondrial function or inhibition of cellular metabolism elicits ER hyperoxidation and delays ER proinsulin export. Our observations have identified a new mechanism linking defects in mitochondrial and redox metabolism with alterations to ER function in the decline of insulin production in T2D. Future goals of this project include: further establishing links between glucose metabolism and ER redox control, defining key mediators that buffer ER redox capacity, and determining how proinsulin folding impacts ER redox homeostasis. For additional information, see our review.
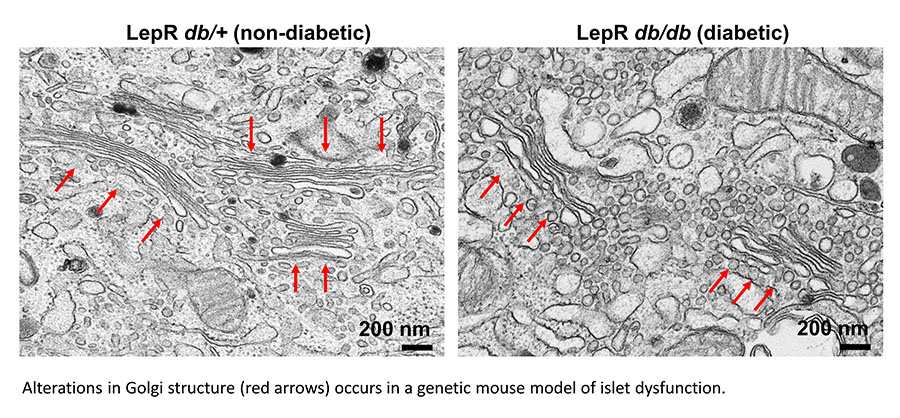
We are also exploring alterations to the Golgi as an underlying mechanism for insulin granule loss in T2D. For example, Golgi cisternae from diabetic animals are distended and highly vesiculated, which may contribute to defects in proinsulin packaging and export. Please see our publication for more information.
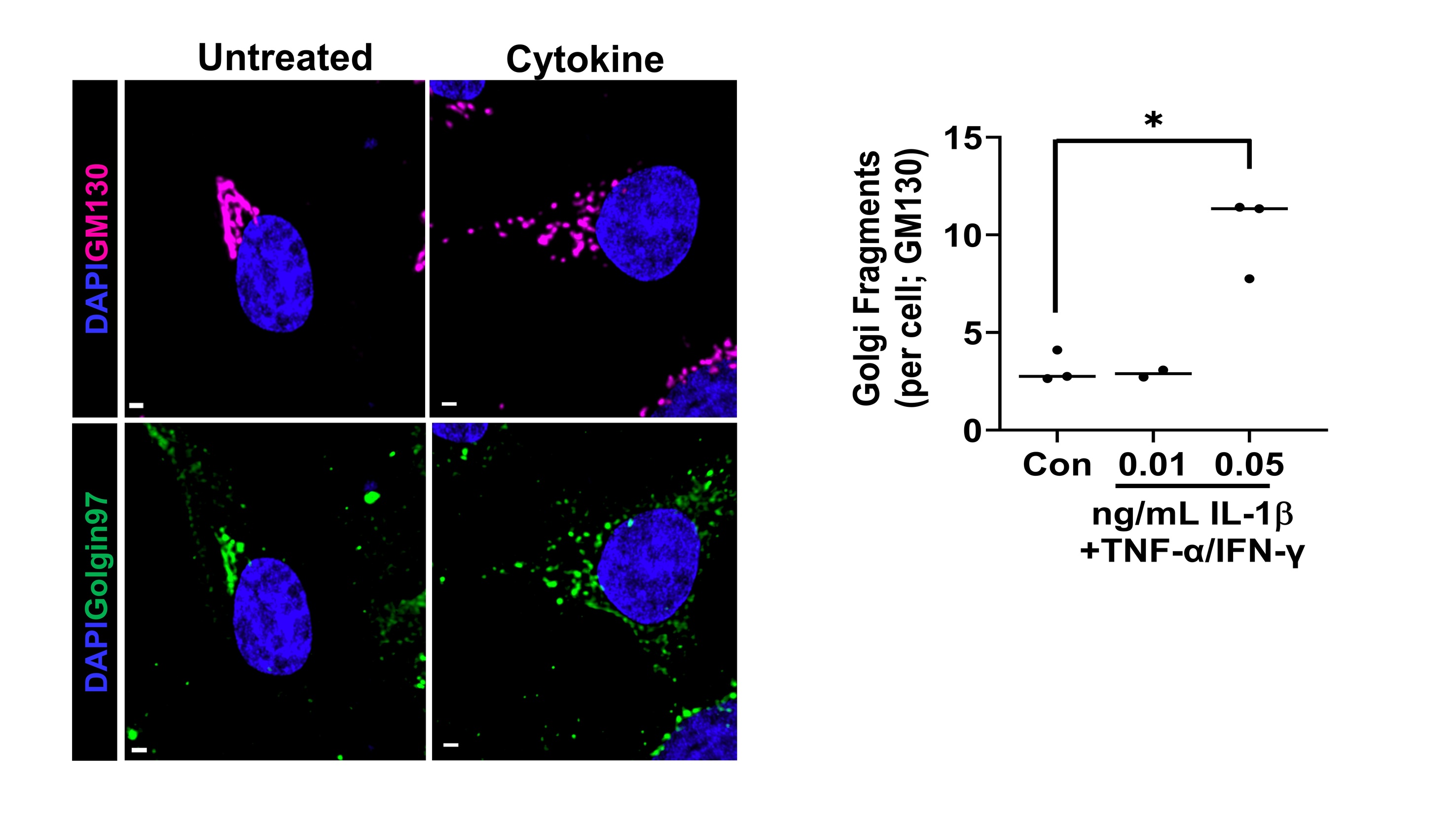
Proinflammatory cytokines alter β-cell Golgi structure
Type 1 Diabetes (T1D) results from the selective autoimmune destruction of insulin-secreting pancreatic β-cells. Emerging evidence highlights the β-cell as an active participant in T1D development, yet how the β-cell contributes to its own demise remains a critical gap in the field. Our recent studies have uncovered a novel role for proinflammatory cytokines in dismantling the β-cell’s secretory program that may directly address this knowledge gap. We have discovered that proinflammatory cytokines elicit a complex change in the β cell’s Golgi structure and function, which has not been previously documented. The structural modifications include Golgi compaction and, in some instances, loss of the inter-connecting ribbon leading to Golgi fragmentation. Please see our publication for more information. Future studies are investigating how cytokines alter Golgi structure and defining the impact of Golgi dysfunction on β-cell surface remodeling.